
掃碼聯系客服


全基因組分析,數十年以來,都是先將測序數據,比對到一個線性的參考序列上,之后進行變異檢測。但當樣品和參考基因組存在較大差異,或種群多樣性較高時,受限于線性的參考基因組帶來的比對偏向性,會影響變異檢測的準確性和敏感性。而采用包含已知變異信息的圖結構做參考基因組,基于此進行變異檢測,被稱為泛基因組,是未來大人群研究的新方向。
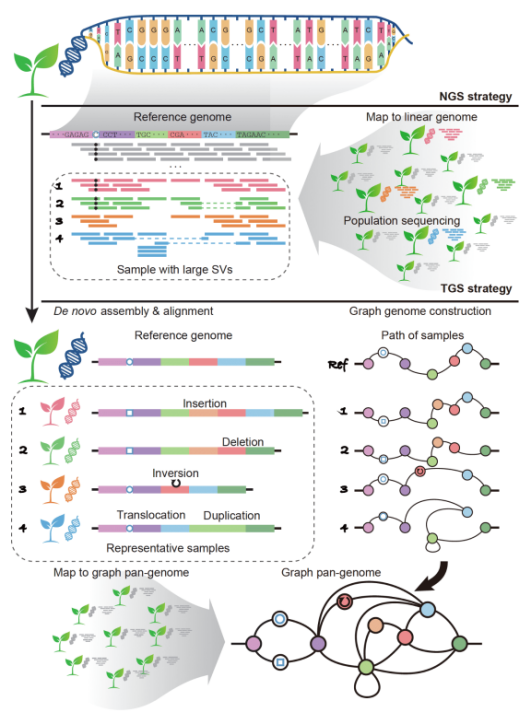
圖參考基因組范例,其中的每個環狀結構,代表種群上的變異
Science12月16日的論文“Pangenomics enables genotyping of known structural variants in 5202 diverse genomes”,介紹了美國加州大學開發的泛基因組比對工具Griaffe,相比常用的bwa,其速度更快,結合變異檢測工具vg,能夠更準確的檢測長度大于50bp的結構變異。

Griaffe的運行速度,相比之前的圖參考序列比對工具,要快一個數量級,比常用的BWA minimap2,也快了50%,見下圖

對于二倍體,雜合變異的兩種堿基,在比對中所支持的比例理論上應該是50%,偏離該比例,說明比對存在偏向性,在低深度時會影響變異檢測的準確性。下圖對比了使用bwa比對到hg19參考基因組和使用vg-map和Griaffe兩種圖比對工具(比對到基于千人變異構建的圖參考序列)之后使用bcftools mpileup和call進行變異檢測后,不同長度的Indel和SNP中,突變型支持的堿基比例。可以看到兩種圖比對工具,在各個長度都接近50%,但bwa比對,隨著變異長度增長,會出現變異型比例下降的趨勢,這與短序列對較長indel的檢出率下降相關。

在標準品HG002上,比較使用不同比對工具,結合Deep variant進行變異堅持得到的結果,左圖比較的是雜合的snp位點,其中使用Griaffe得出的結果,假陽性和假陰性都更低。而右圖則是比較長于50bp的插入刪除的準確性,可以看到使用Griaffer后,F1在全基因范圍和高可信度范圍,能達到0.9左右,說明Griaffe能以更快的速度,達到和之前的圖比對工具在結構變異檢測上相同的精度。

之前的全基因組研究,受限于方法學,多關注單堿基變異或短的插入刪除,而對結構變異,缺少準確且全面的檢測方法。該論文通過使用三代長讀長序列檢測到的變異為金標準,對不同人群的5202人(包含全部千人基因組的樣本)的結構變異進行檢測,指出使用泛基因組的工具,可基于短序列,對結構變異進行更精準的檢測。
具體流程如下圖,先從比對生成的cram文件中,結合圖參考基因組的index文件,拿到原始的read文件,之后使用Griaffe進行比對,再進行結構變異的變異檢測,其中大部分的計算,都花在比對上,單樣本全流程的核時數在206小時,相比傳統的全基因組分析更快。右圖比較了使用不同的圖參考基因組后,長的插入和刪除的平均F1值,相比之前的結構變異檢測更準。

對得出的結構變異,進行PCA分析,可以得出和snp類似的結果,見下圖,這意味著在人群中,不同種族在結構變異上存在著頻率差異,例如VNTR基因上的變異,在非洲人群中出現的比例超過14%,而在其它人群中低于3%。

Griaffe的安裝和使用都很簡單,github(https://github.com/vgteam/vg)上有編譯好的二進制文件,下載后可在linux平臺直接運行。之后可以基于參考基因組和變異的vcf文件,構建圖參考序列的index文件,推薦選擇和研究人群相近的群體,或者多樣性高的群體變異信息(需包含每個個體的變異情況)
命令如下:
vg construct -r small/x.fa -v small/x.vcf.gz >x.vg
構建好的圖參考序列,可以進行可視化,或壓縮為二進制文件節省存儲。
之后的比對過程,先生成后綴為gam的圖比對結果,之后轉換為常用的bam文件,可進行接下來的變異檢測,命令如下:
vg map -T x.sim.txt -x x.xg -g x.gcsa > aln.gam
vg surject -x x.xg -b aln.gam > aln.bam
新檢出的樣本上的變異,還可以用來擴展參考序列的多樣性,即疊加式的構建更準確的圖參考基因組,該操作的輸入是圖參考序列和比對生成對的.gam文件,輸出是增強后的參考序列,對應的命令是;
vg augment x.vg aln.gam -i -S > aug_with_paths.vg
之后的變異檢測,可以基于bam文件,使用傳統的Deep variant這樣的工具,也可以使用vg,以.gam文件為輸入,通過以下命令得到檢測結果:
vg pack -x x.xg -g aln.gam -Q 5 -o aln.pack
vg call x.xg -k aln.pack > graph_calls.vcf
對于多樣本的情況,可以使用bcftools merge,對檢出的變異進行合并。對于結構變異,該研究中將刪除或插入位置在20bp之內,且序列相似度在80%以上的變異,在群體上合并,據此計算人群頻率,通過RepeatMasker進行功能注釋。
類似基于比對結果,對參考基因組進行增強,將新發現的變異加入參考基因組中,vg也支持輸入為vcf格式的圖參考基因組增強,或使用局部組裝得出的長序列,通過vg mpmap命令比對到參考基因組上,構建描述結構變異多樣性的圖參考序列,之后使用vg concat命令,將圖參考基因組合并,得到用于結構檢測檢測的圖參考序列。
對于轉錄組數據,也可以使用vg進行比對,從而應對不同的可變剪切帶來的比對偏向性。先是基于線性參考基因組,變異信息,和注釋文件,構建圖結構的index
vg autoindex --workflow mpmap -t 4 --prefix vg_rna --ref-fasta small/x.fa --vcf small/x.vcf.gz --tx-gff small/x.gtf
之后進行比對
vg mpmap -n rna -t 4 -x vg_rna.spliced.xg -g vg_rna.spliced.gcsa -d vg_rna.spliced.dist -f small/x_rna_1.fq -f small/x_rna_2.fq > mpmap.gamp
泛基因組分析,是大規模人群基因組未來的發展方向,不論對于人群,動植物還是微生物,使用包含變異信息的圖參考序列,都能更好的應對基因多樣性帶來的偏向性。通過引入高性能圖基因組比對工具griaffe,降低了使用圖參考序列的門檻。具體首先體現在之前的比對工具,相比bwa慢4-5倍,而新工具比bwa更快,其次是通過支持將圖參考序列的比對結果轉換為bam格式,使得現有的變異檢測等分析工具可兼容使用。新研究還通過了大規模人群數據,說明了短序列使用圖參考序列,可以得出準確的結構變異檢測結果,其人群變異攜帶頻率和之前研究相符合。最后,vg還可以基于從頭組裝的結果,構建圖參考基因組,隨著未來de novo組裝研究的增多,這將取代基于變異信息構建的圖參考序列,稱為泛基因組研究的新方向。




